The 2019 pandemic of coronavirus infection (COVID-19) has been raiding the globe for more than a year, with more and more new concerns emerging that weaken conventional measures aimed at controlling the spread. The health care systems of many countries are struggling to deal with urgent cases caused by new changes with higher transmission observed of the true respiratory coronavirus 2 (SARS-CoV-2) syndrome, the virus causing COVID-19.
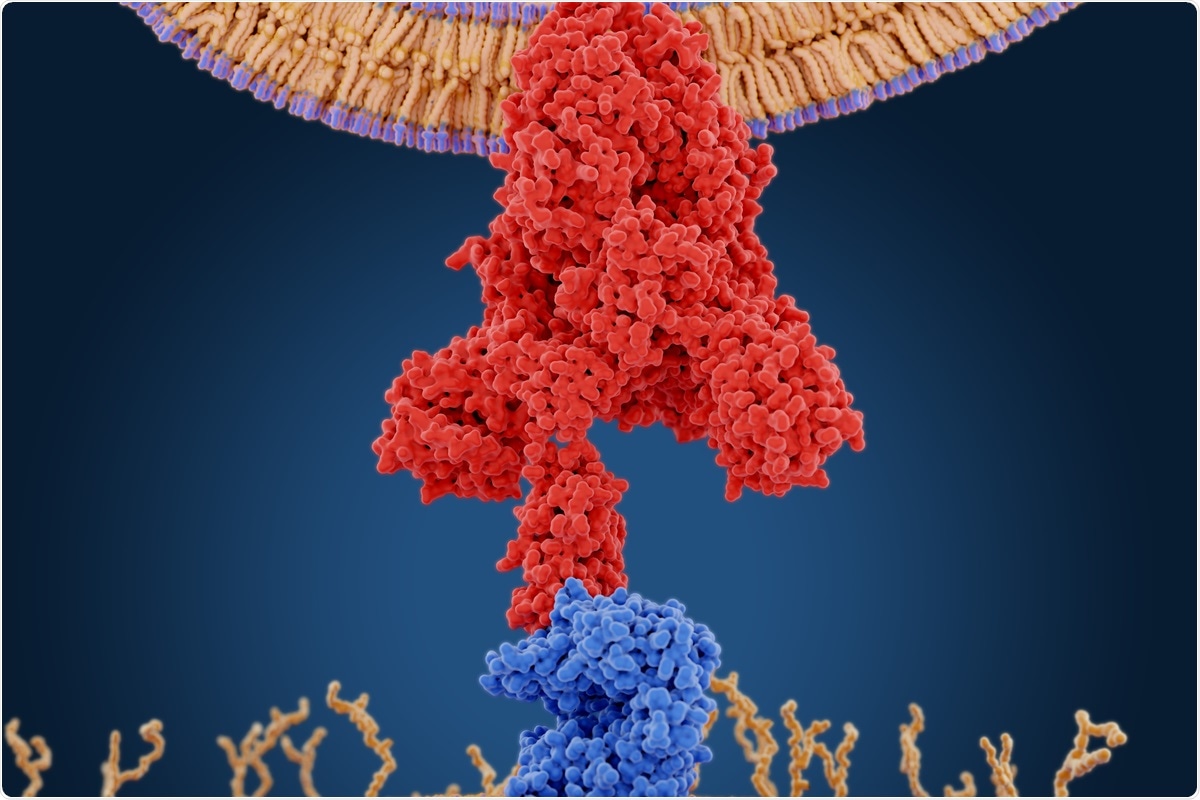
Monitoring changes in SARS-CoV-2 represents a major challenge in the current global health crisis, as well as future viral outbreaks. In particular, mutations and deletions in SARS-CoV-2 spike proteins have significant effects on vaccines and drugs that target this major structural viral protein to stop the pandemic.
Now, researchers in the U.S. – at Virginia Commonwealth University, the University of Minnesota, and NASA’s Ames Research Center – have shown how maps of dynamic landscapes or points of call, along with alignment information, can series, identify major residue mutations and spike proteins. deletions linked to emerging changes.
SARS-CoV-2 mutations
Since SARS-CoV-2 first appeared in Wuhan City, Hubei Province, China, in December 2019, it has shown high transmission rates worldwide. To date, the virus has spread to 192 countries and regions, causing more than 107.48 million cases and 2.35 million deaths.
One of the current concerns about pandemic is that the virus is still circulating. Emerging changes show higher levels of transmission and infectivity, therefore, challenging routine and improving vaccines. The SARS-CoV-2 conversion rate is estimated at ∼ 10−3 substitution per site per year.
In the past, the largest protein mutation of SARS-CoV-2 is the D614G spike protein mutation, which has been linked to viral loads of the upper respiratory tract. The mutation is thought to be ubiquitous in recent genomic sequences around the world. In addition, another variable with the variable name RA or VOC 202012/01 or B.1.1.7 has been identified over the past few months. The variant is more contagious, and there are both deletions and mutations in the spike protein (including D1614G).
It is essential to find out how current and future changes lead to altered transmission rates, viral loading differences, protection against antibody and vaccine, and against enhanced drugs for SARS-CoV-2.
The study
In the study, which appeared on the pre-print medRxiv * server, the team focused on the analysis of mutations of the spike protein, as a partial guide to the potential effects of its mutations on viral function, structure, and potential behavioral changes.
The team also found an excellent homology of stabilizing residual glue points in spike coronavirus proteins, called series homologous glue points. Overall, these mutations or deletions of latent residues were evaluated using all-atomic biocomputational molecular dynamics over approximately one microsecond to ensure structural and volatile changes in the variables associated with spike proteins.
The team also specifically studied both the triple mutant based on SARS-CoV-2 theory and the RA or B.1.1.7 version. They analyze the structure and main dynamic motions associated with the Down versus Up protomer states of the virus, which is the triple mutant that can interact with a neighboring N-terminal land-binding (RBD-NTD) weaken.
In addition, the researchers conducted a comprehensive study of the UK variant and D614G mutation to find key differences in protomer configuration, which may affect the efficacy of vaccines and drugs to treat pandemic.
Using the same method, the team analyzed the UK variable to find major mutations or deletions in its spike protein, which may be responsible for the difference being more pronounced. movement.
The team’s decisions resulted in two major modifications, the D614G and the N501Y. Biochemical tests confirmed changes associated with the D614G that make it to an accessible state of angiotensin-converting enzyme 2 (ACE2). At the same time, the N501Y has a 41Y human ACE2 call point partner, which could lead to a strong residual pair interaction. This may explain why South Africa has a higher disease rate of the UKB1.1.7 and B.1.351 changes.
The team concluded that these two major mutations could promote transmission and infection rates in two different ways. However, the team confirmed that further studies are needed to measure this feature and its outcome.
* Important message
bioRxiv publish preliminary scientific reports that are not peer-reviewed and, therefore, should not be seen as final, guiding health-related clinical practice / behavior, or be treated as information established.
Magazine Reference:
- Peters, M., Bastidas, O., Kokron, D., and Henze, C. (2021). Transformations, Comparisons, and Analysis of Protomer States Down to Up Transformations of SARS-CoV-2 Spike Prefusion Proteins Including RA Variant B.1.1.7. bioRxiv. doi: https://doi.org/10.1101/2021.02.09.430519, https://www.biorxiv.org/content/10.1101/2021.02.09.430519v1